git clone https://github.com/at-cg/minichain
cd minichain && makeHaplotype-aware alignment of a sequence to a pangenome graph (GFA v1.1)
For this use-case, the reference haplotypes must be stored as paths in the input GFA file.
# Map sequence to haplotype-aware pangenome graph with recombination penalty(R) 10000
./minichain -cx lr test/Graphs/C4-CHM13.gfa test/Genomes/C4-HG03492.2.fa -R10000 > C4-HG03492.2.gafIf the graph does not specify haplotype paths, Minichain uses haplotype-agnostic chaining algorithm.
# Map sequence to pangenome graph
./minichain -cx lr test/Graphs/C4-CHM13_mg.gfa test/Genomes/C4-HG03492.2.fa > C4-HG03492.2.gafMinichain is a haplotype-aware sequence aligner to a pangenome graph represented as DAGs. It can scale to pangenomes built from several human genome assemblies. We have implemented two provably-good algorithms:
- Gap-sensitive co-linear chaining algorithm (GFA v1.0, rGFA).
- Haplotype-aware co-linear chaining algorithm (GFA v1.1).
Please refer to our publications for details about the algorithms.
These algorithms enable accurate and fast alignments of long reads or phased contigs. Minichain borrows seeding and base-to-base alignment code from Minigraph.
git clone https://github.com/at-cg/minichain
cd minichain && make
# Check installation
./minichain --versionMinichain can be used for both sequence-to-sequence alignment as well as sequence-to-graph alignment. A graph should be provided in either GFA v1.0, rGFA or GFA v1.1 (haplotype-aware) format. Minichain automatically uses either haplotype-aware or haplotype-agnostic chaining algorithm depending on whether the haplotype paths are stored in the input pangenome graph.
Users can run quick tests on sample data using the following commands. The alignment output is provided in either PAF or GAF format.
# Map sequence to sequence
./minichain -cx lr test/Genomes/C4-CHM13.fa test/Genomes/C4-HG03492.2.fa > C4-HG03492.2.paf
# Map sequence to haplotype-aware pangenome graph with recombination penalty(R) 10000
./minichain -cx lr test/Graphs/C4-CHM13.gfa test/Genomes/C4-HG03492.2.fa -R10000 > C4-HG03492.2.gaf
# Map sequence to pangenome graph
./minichain -cx lr test/Graphs/C4-CHM13_mg.gfa test/Genomes/C4-HG03492.2.fa > C4-HG03492.2.gaf
Minichain can be used for the incremental graph generation. Sequences should be provided in FASTA format. Users can run quick tests on sample data using the following command. The graph is produced in rGFA format.
# Incremental graph generation
./minichain -cxggs test/Genomes/C4-CHM13.fa test/Genomes/C4-HG002.1.fa test/Genomes/C4-HG002.2.fa > C4-CHM13.gfaWe benchmarked Minichain (v1.3) using simulated queries from a MHC pangenome graph. We simulated each query as an imperfect mosaic of the reference haplotypes. Our results show that haplotype-aware co-linear chains are more consistent with the true recombination events as compared to haplotype-agnostic (recombination penalty = 0) and haplotype-restricted (recombination penalty = ∞). The scripts to reproduce this benchmark are available here.
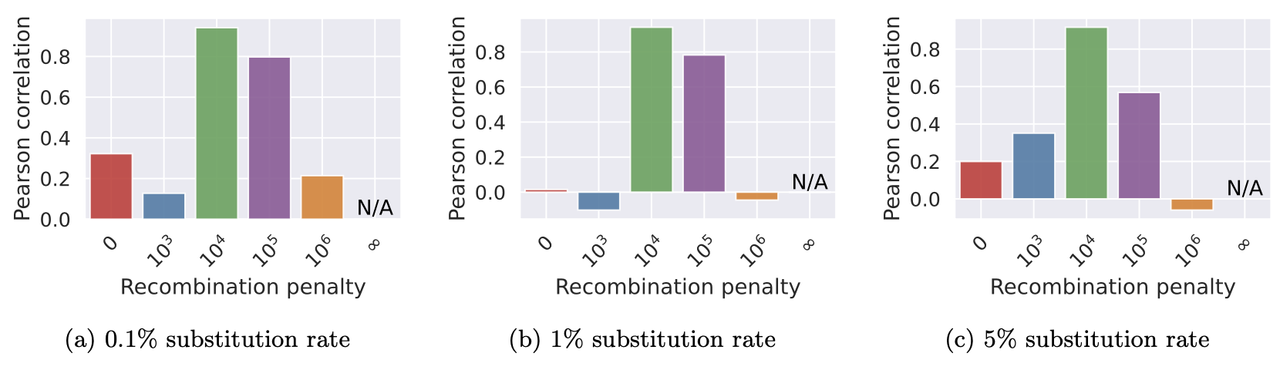
Pearson correlation between the count of recombinations in Minichain’s output chain and the true count.
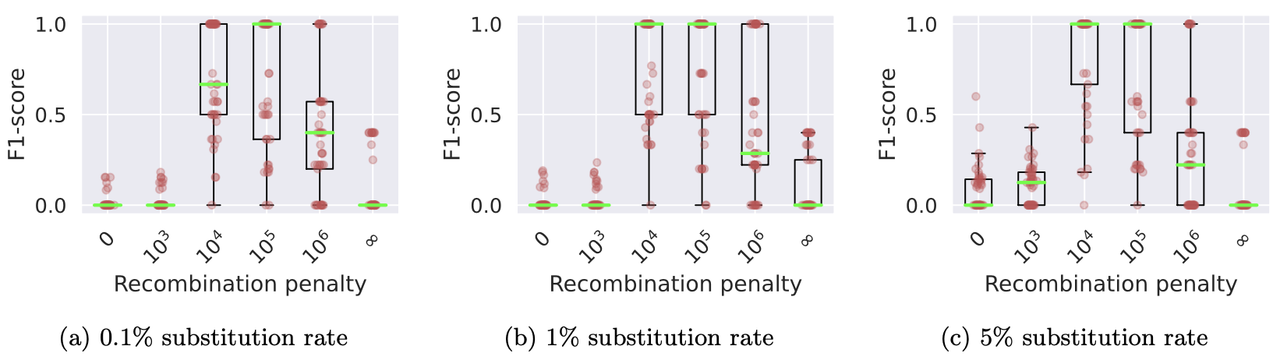
Box plots show the levels of consistency between the haplotype recombination pairs in Minichain’s output chain and the ground-truth. We used different substitution rates and recombination penalties. Median values are highlighted with light green lines.
Datasets for benchmarking are available at Zenodo
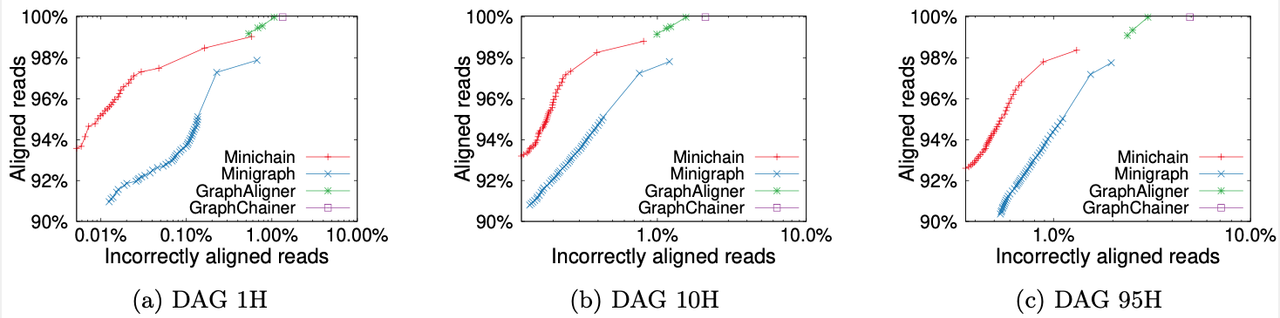
We compared Minichain (v1.2) with existing sequence to graph aligners to demonstrate scalability and accuracy gains. Our experiments used human pangenome DAGs built by using subsets of 94 high-quality haplotype assemblies provided by the Human Pangenome Reference Consortium, and CHM13 human genome assembly provided by the Telomere-to-Telomere consortium. Using a simulated long read dataset with 0.5x coverage, and DAGs of three different sizes, we see superior read mapping precision (as shown in the figure). For the largest DAG constructed from all 95 haplotypes, Minichain used 10 minutes and 25 GB RAM with 32 threads. The scripts to reproduce this benchmark are available here.
Real dataset: We benchmarked Minichain (v1.2) for mapping the UL ONT (#reads: 13589524, N50: 52464) reads from the Human Pangenome Reference Consortium with approximately 52X total coverage to the largest DAG constructed from all 95 haplotypes. Minichain took 13 hours and 28 minutes, utilizing 66 GB of RAM and 128 physical cores (Perlmutter cpu node) and aligned 86% of the sequencing throughput.
Graph generation: Minichain (v1.1) can construct a human pangenome graph. Our experiments utilized 94 high-quality haplotype assemblies from the Human Pangenome Reference Consortium and CHM13 human genome assembly from the Telomere-to-Telomere consortium. Minichain took 58 hours and 17 minutes, utilizing 483 GB of RAM and 32 threads (Cori Large Memory node).
We plan to continue adding features in future releases.
- Support for graphs with SNPs and indels.
- Support for haplotype-aware graphs constructed using fragmented assemblies.
- Support for haplotype-aware extension (base-to-base alignment).
- Support for cyclic pangenome graphs.
- Ghanshyam Chandra and Chirag Jain. "Haplotype-aware Sequence-to-Graph Alignment". RECOMB 2024.
- Ghanshyam Chandra and Chirag Jain. "Sequence to Graph Alignment Using Gap-Sensitive Co-linear Chaining". RECOMB 2023.