Home
Maintainers: Mike Lin and Irwin Jungreis
PhyloCSF is a method to determine whether a multi-species nucleotide sequence alignment is likely to represent a protein-coding region. PhyloCSF does not rely on homology to known protein sequences; instead, it examines evolutionary signatures characteristic to alignments of conserved coding regions, such as the high frequencies of synonymous codon substitutions and conservative amino acid substitutions, and the low frequencies of other missense and non-sense substitutions (CSF = Codon Substitution Frequencies). One of PhyloCSF’s main current applications is to help distinguish protein-coding and non-coding RNAs represented among novel transcript models obtained from high-throughput transcriptome sequencing.
The mathematical details of the method, and benchmarks comparing it to other approaches, are described in:
- Lin MF, Jungreis I, and Kellis M (2011). PhyloCSF: a comparative genomics method to distinguish protein-coding and non-coding regions. Bioinformatics 27:i275-i282 (ISMB/ECCB 2011)
The following papers also provide relevant background and/or illustrate potential applications.
- Lin MF, Carlson JW, Crosby MA, Matthews BA, et al. (2007). Revisiting the protein-coding gene catalog of Drosophila melanogaster using twelve fly genomes. Genome Research 17:1823-1836
- Lin MF, Deoras AN, Rasmussen MD, and Kellis M (2008). Performance and scalability of discriminative metrics for comparative gene identification in 12 Drosophila genomes. PLoS Comput Biol 4:e1000067
- Guttman M, Garber M, Levin JZ, Donaghey J, et al. (2010). Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotech 28: 503-510
The remainder of this document discusses how to obtain and use the publicly available implementation of PhyloCSF. We keep a WishList of planned or (mostly) wished-for future improvements.
Input: The user must prepare a cross-species nucleotide sequence alignment for PhyloCSF. PhyloCSF can be applied to alignments of complete open reading frames (ORFs), individual exons, or complete or partial transcripts (i.e. concatenated exons).
Output: PhyloCSF outputs a score, positive if the alignment is likely to represent a conserved coding region, and negative otherwise.
As an alternative to installing and running the PhyloCSF software, PhyloCSF scores for selected phylogenies may be viewed in the UCSC Genome Browser by copying the URL “http://www.broadinstitute.org/compbio1/PhyloCSFtracks/trackHub/hub.txt” into the “My Hubs” tab under “track hubs”, or in the BioDalliance browser by adding that track hub on the GENCODE web site. Detailed explanation of the tracks can be found here.
PhyloCSF works on Linux and Mac OS X. If you like to work with Docker, Dockerfiles are available (basic, advanced). Otherwise, follow these steps:
This will install a minimal OCaml toolchain (if not already present on your system) and the OPAM package manager. Once it’s installed, initialize the installation by running opam init and then set up your environment with eval $(opam config env).
opam install batteries ocaml-twt gslTo successfully install the gsl package, which provides OCaml bindings to the GNU Scientific Library (GSL), you may also need to install GSL itself, if it’s not already available on your system. On Debian/Ubuntu systems GSL is available in the libgsl0-dev package, and on Mac OS X, gsl is available in both Homebrew and MacPorts. Once GSL itself is installed, it’s still necessary to install the OCaml bindings using opam, as shown above.
git clone git://github.com/mlin/PhyloCSF.git
cd PhyloCSF
makeOnce the software compiles successfully, you can then run ./PhyloCSF as in the examples shown below.
PhyloCSF should be usable in this way by anyone on the machine/cluster with rx permissions; they don’t have to set up OPAM or anything else. PhyloCSF can be started from any working directory.
If you want to “install” PhyloCSF somewhere, you must transplant ./PhyloCSF, ./PhyloCSF.ARCH (where e.g. ARCH=Linux.x86_64), and the ./PhyloCSF_Parameters directory to the desired location.
To run PhyloCSF’s tests, first additionally opam install ounit should. Then make test to run the full suite, which can take 5-10 minutes, or SKIP_SLOW=1 make test to run a <1-minute subset. The tests also run on Travis CI:

PhyloCSF leaves it up to you to extract and align the orthologous loci from the genomes you wish to use. Galaxy and PHAST both have some tools that can help you extract and “stitch” blocks from MAF whole-genome multiple alignments. Alternatively, you might cook up your own alignments of mRNA sequences from different species (see the Aldh2 example below).
The input alignments for PhyloCSF need to be in FASTA format. Here’s an example, representing the complete ORF of tal-AA in Drosophila melanogaster, aligned with the orthologous regions from D. ananassae, D. pseudoobscura, D. willistoni, and D. virilis.
PhyloCSF_Examples/tal-AA.fa
>dmel|chr3R:9,639,593-9,639,688| ATGCTGGATCCCACTGGAACATACCGGCGACCACGCGACACGCAGGACTCCCGCCAAAAGAGGCGACAGGACTGCCTGGATCCAACCGGGCAGTAC >dana ATGCTGGATCCCACAGGAACGTACAGGCGACCCCGCGACTCGCAGGACACACGCCAGAAGCGGCGCCAGGATTGCCTGGATCCAACCGGGCAGTAC >dpse ATGCTGGATCCCACAGGAACCTACCGTCGCCCACGCGACACAAAAGACACACGCCAGAAACGGCGTCAGGACTGCCTAGATCCCACCGGGCAGTAC >dwil ATGCTGGATCCAACTGGTACATATCGCCGACCAAGGGATACACAGGACACACGCCATAAACGGC---------GCCTGGATCCAACTGGACAATAT >dvir ATGCTGGATCCAACGGGCACCTATCGGCGGCCGCGTGAGTCGCGTGACACGCGCCACAAGCAGCGGCAGCTGGCGCTGGATCCTACCGGGCAGTAC
Details:
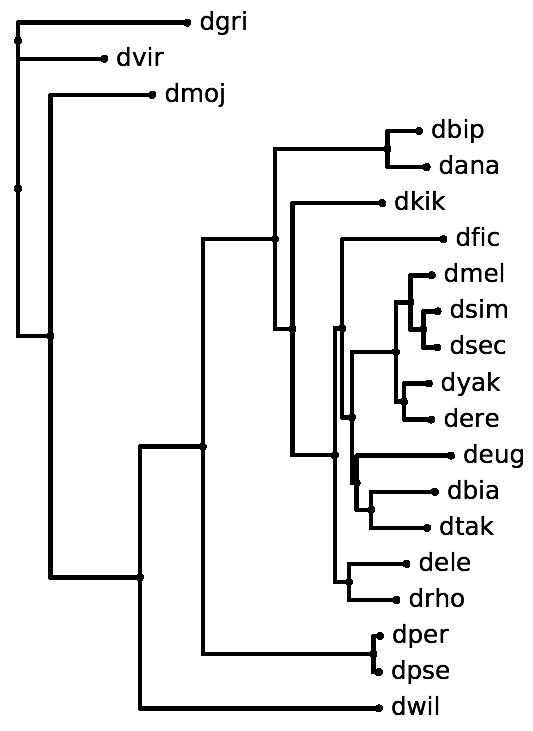
- The alignment may represent anywhere from two up to all of the available species in one of the phylogenies for which parameters are available (see below). For example, we provide parameters for twelve Drosophila species but the above alignment includes only five.
- By default, the reference sequence (first row) must be ungapped, to ensure the reading frame is completely unambiguous. This can, however, be relaxed using command-line options.
- The order of the rows does not matter, as long as the species name in each header line can be matched up with the leaves of the phylogenetic tree in the parameters. In particular, any available species can be used as the reference sequence, as long as it comes first.
- Any gapped codons are marginalized out (standard for phylogenetic likelihood calculations)
- Everything including and after the first | (pipe) in a header line is ignored.
- The sequences can contain newlines but not any other whitespace.
- The program will pretend U’s are T’s.
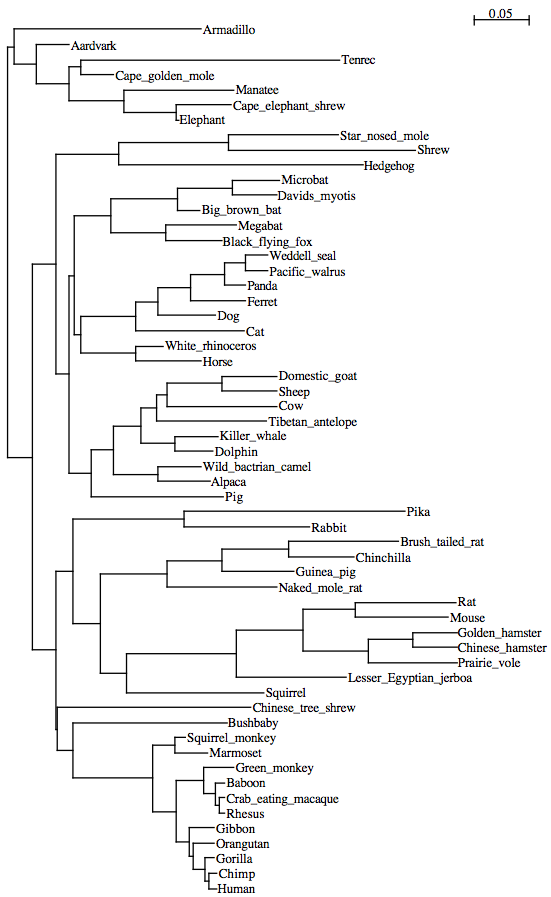
29mammals
58mammals
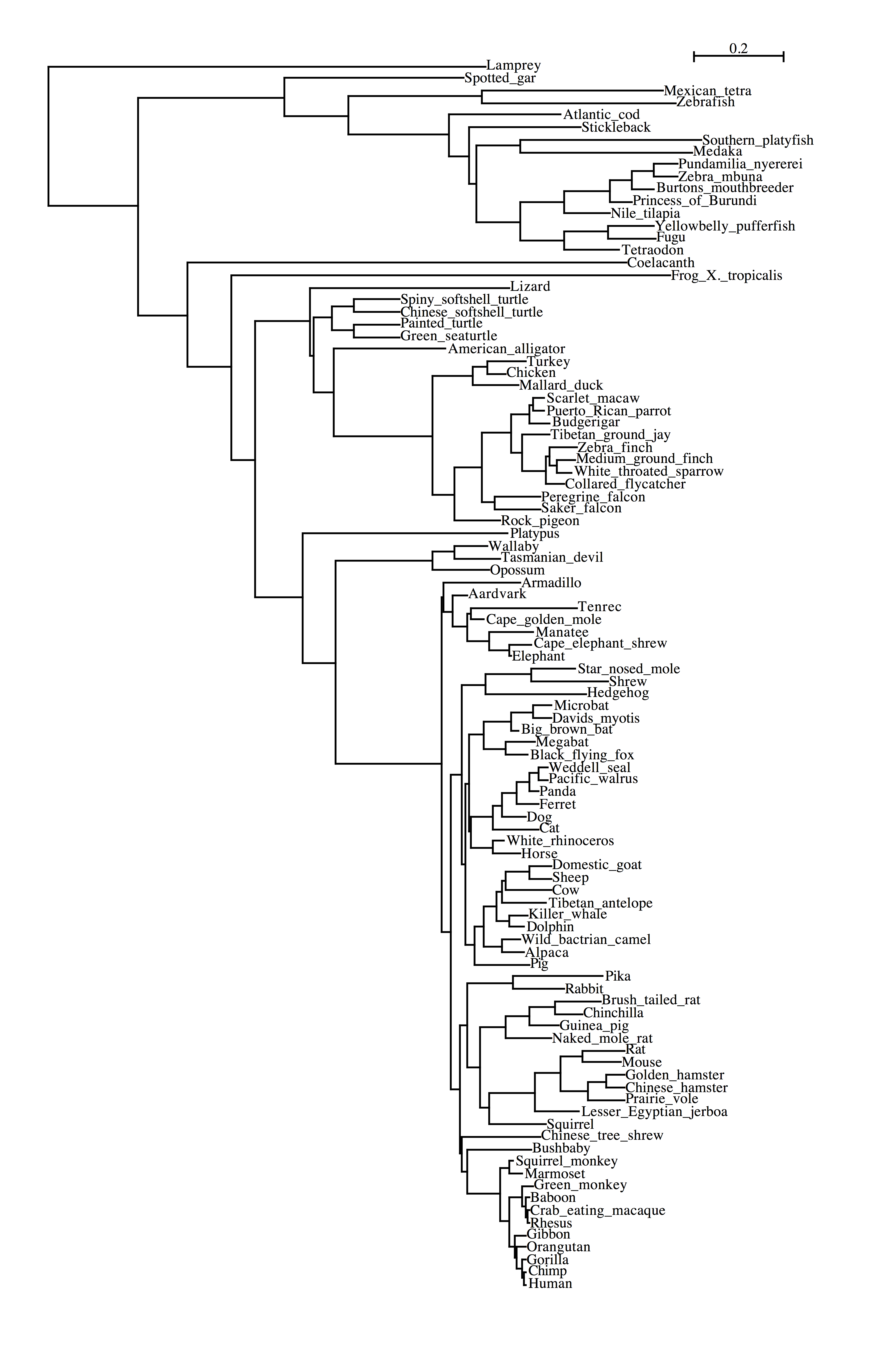
100vertebrates
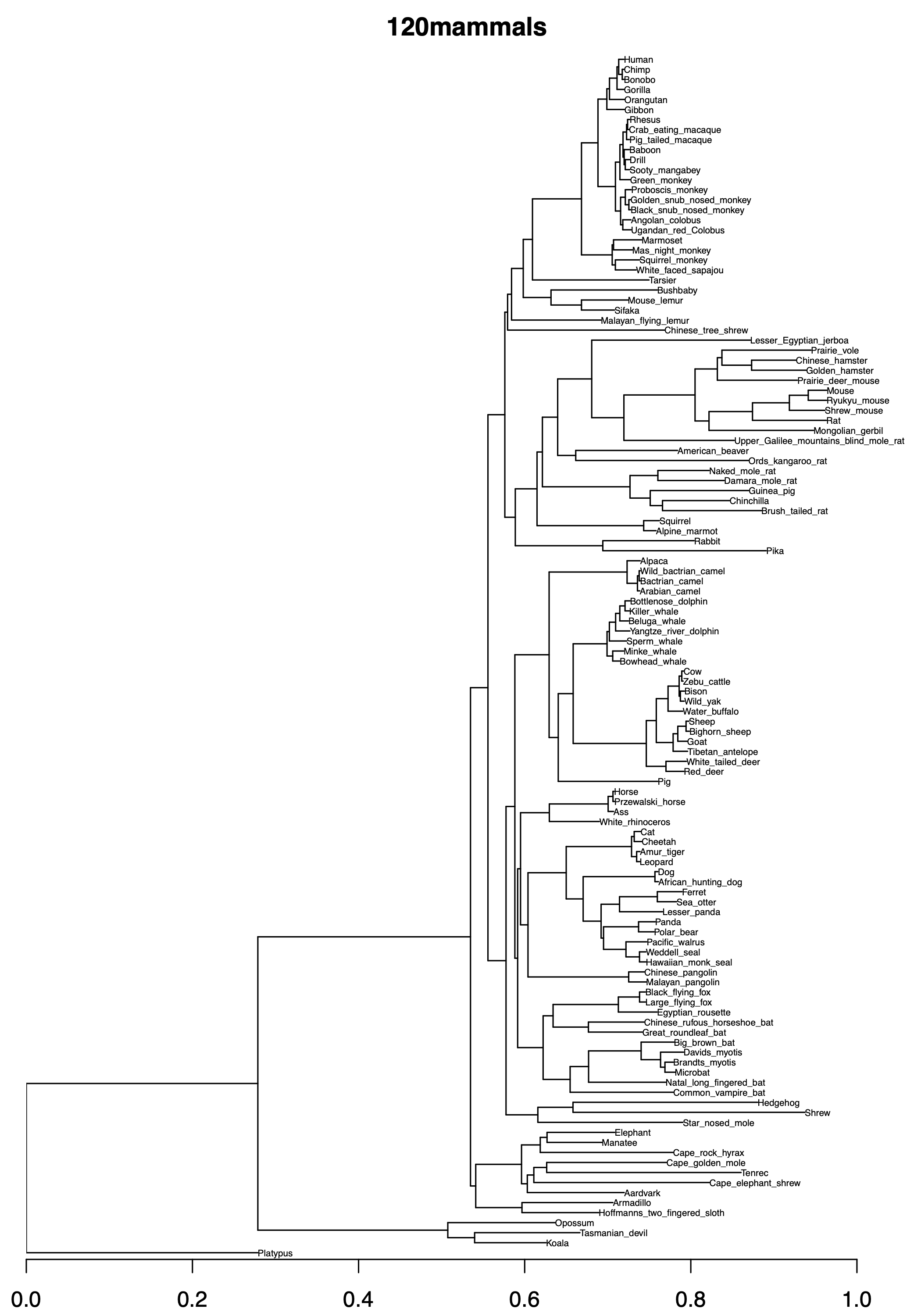
120mammals
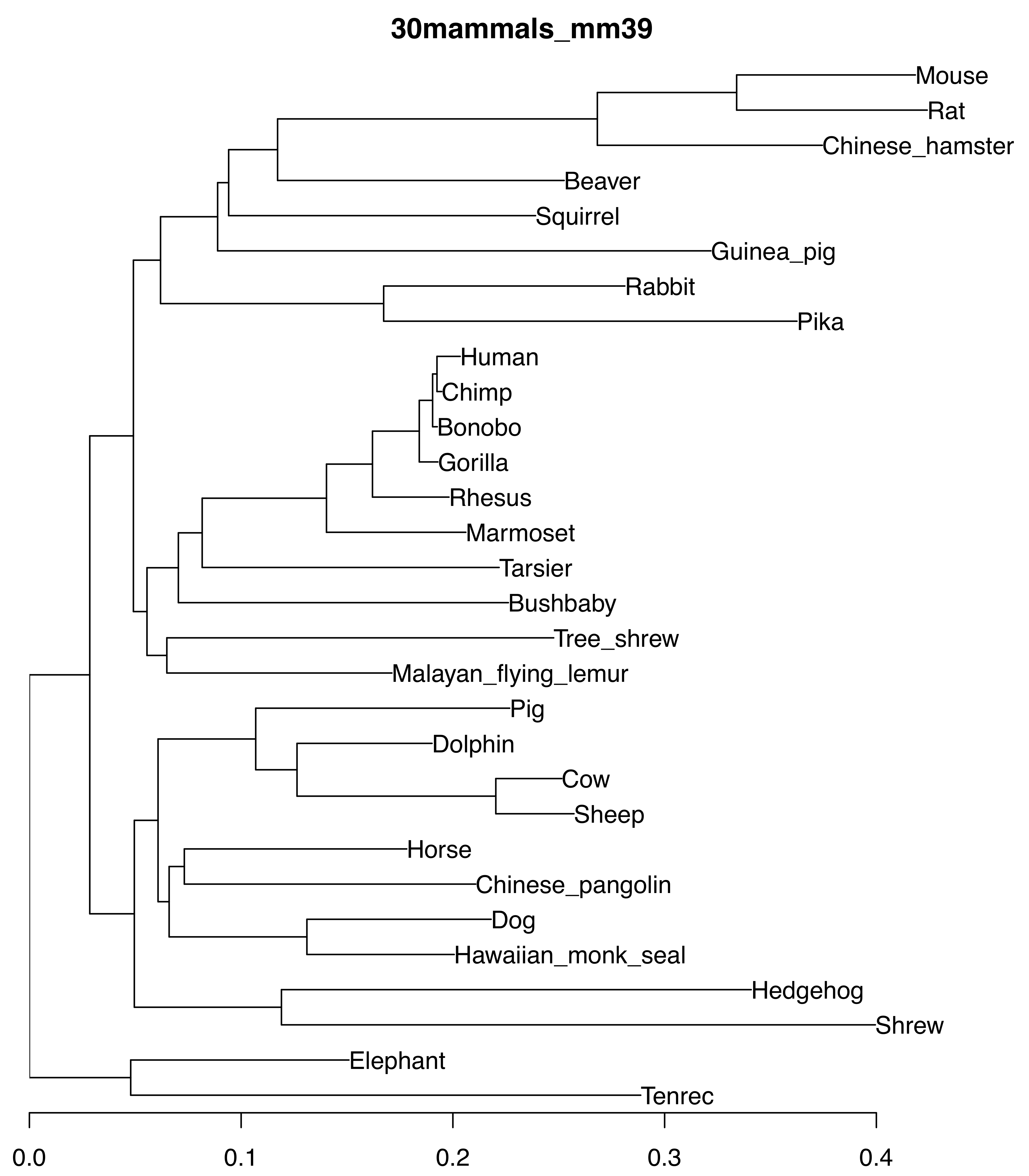
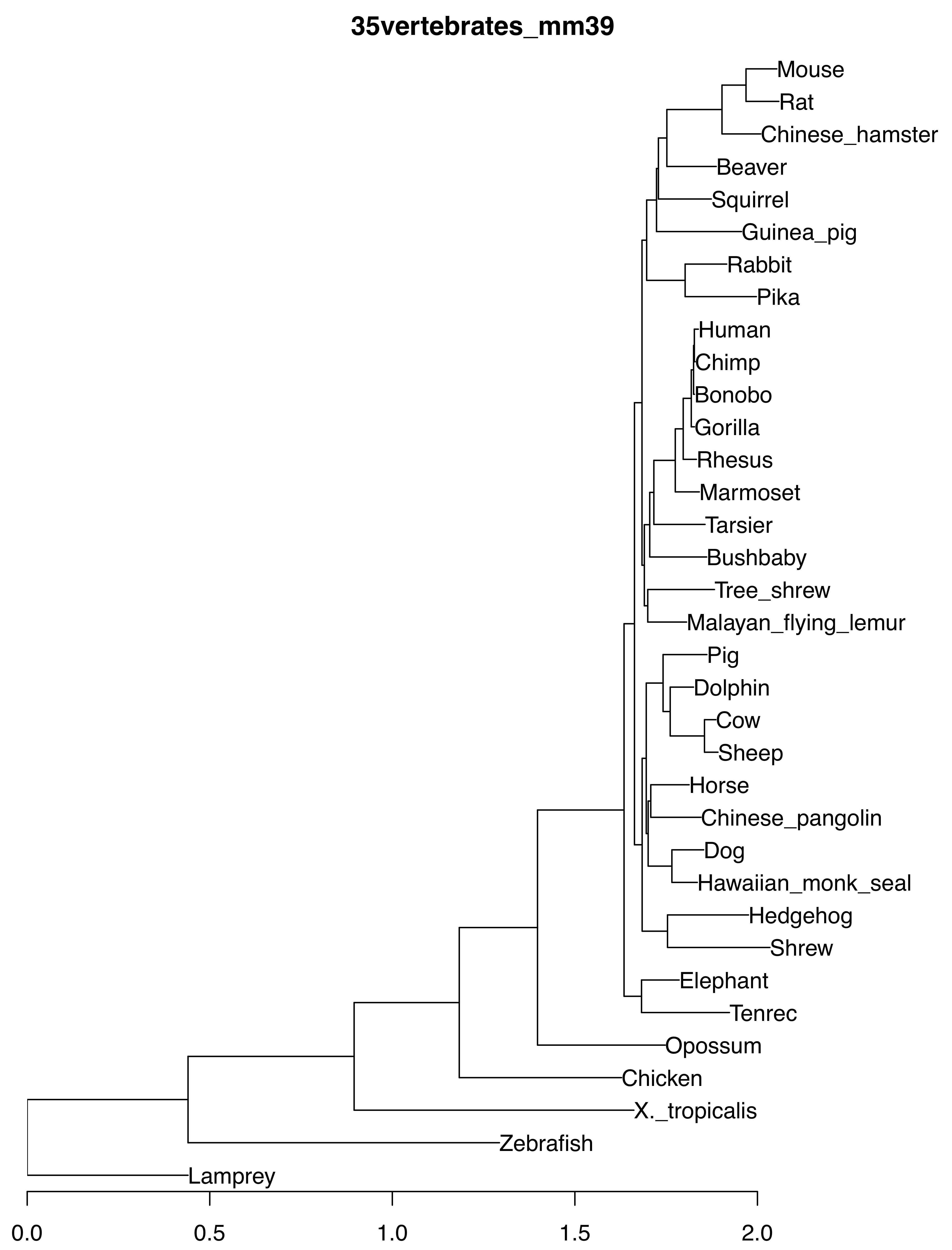
30mammals_mm39
35vertebrates_mm39
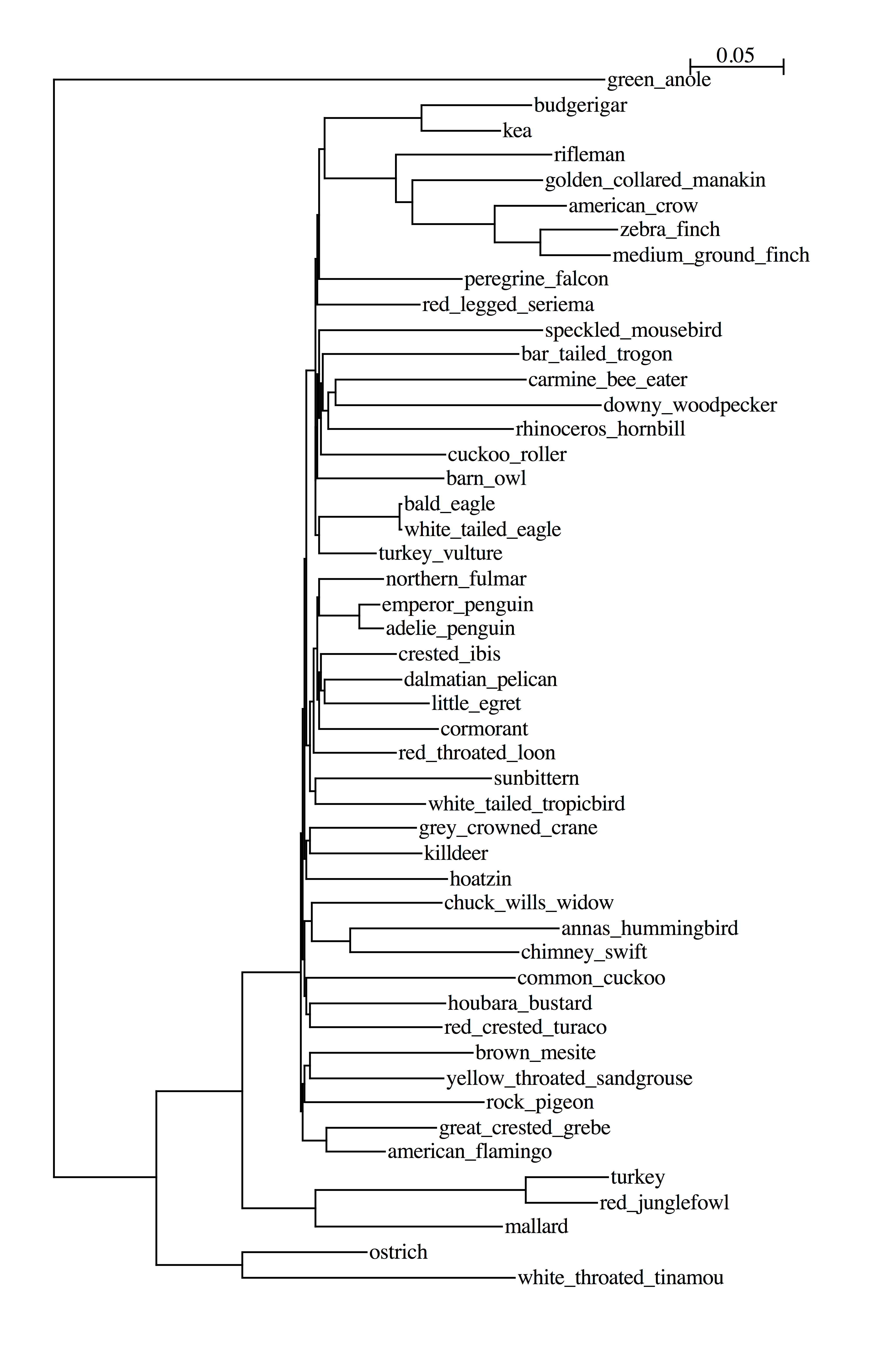
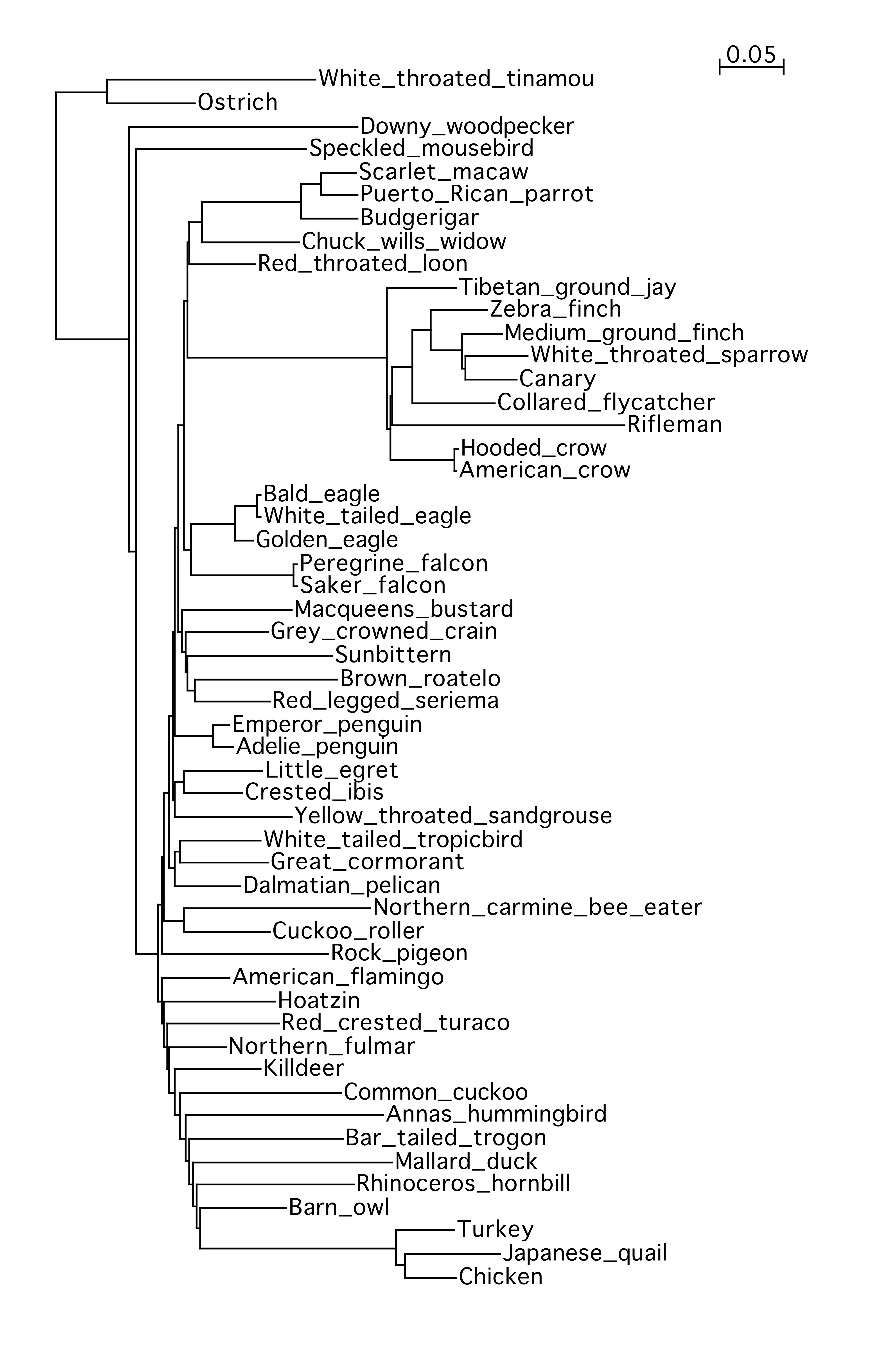
49birds
53birds
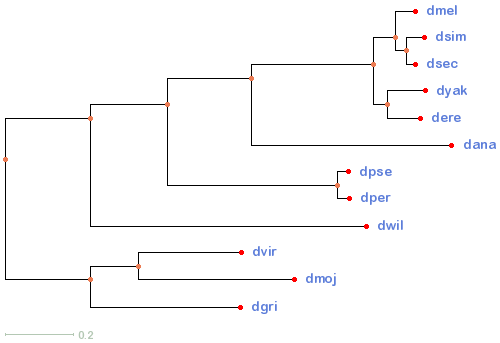
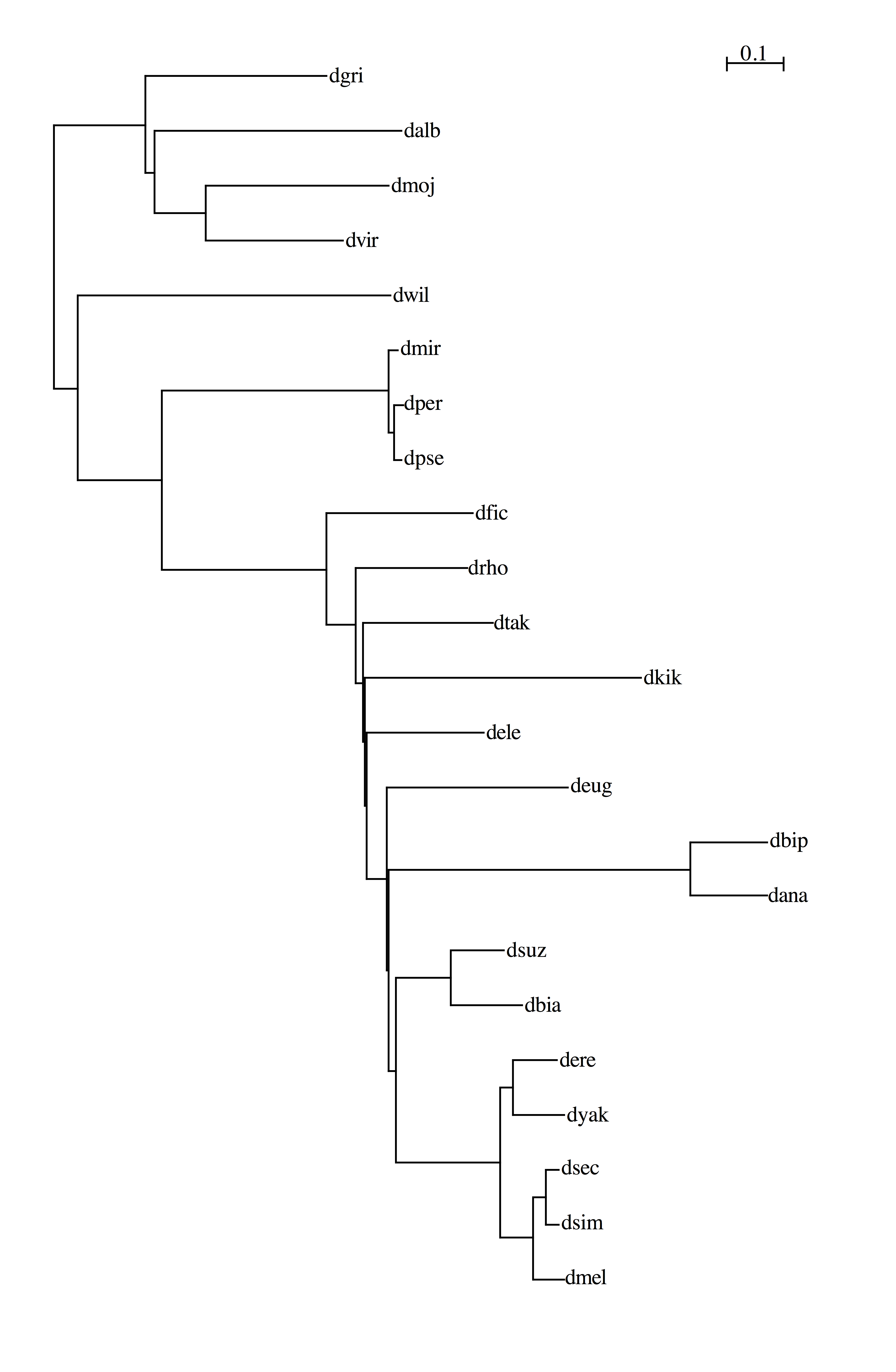
12flies
20flies
23flies
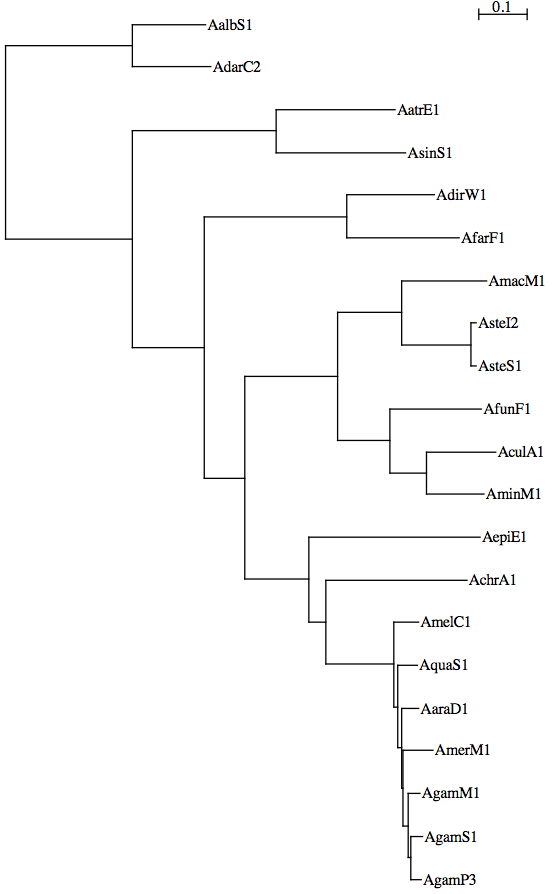
21mosquitoes
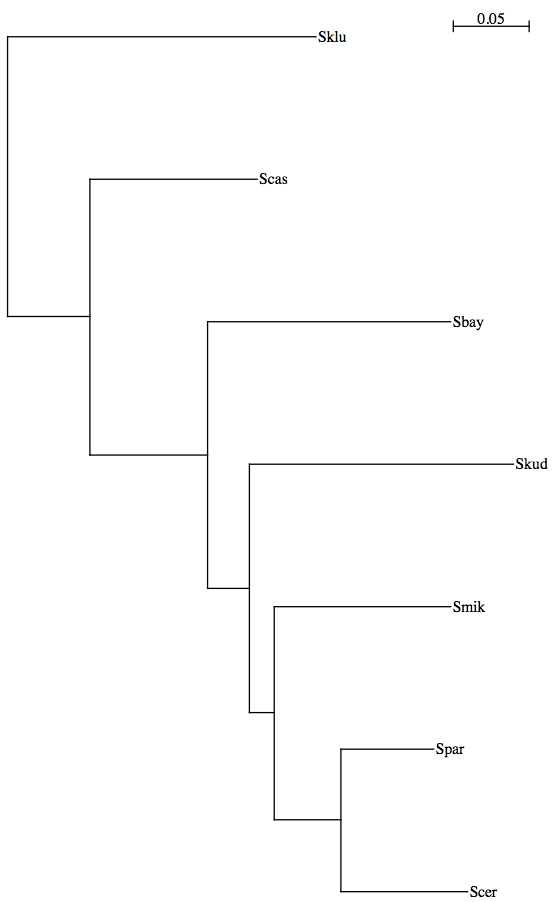
7yeast
26worms
Although your alignments may provide any subset of the species in these phylogenies, some subsets are more useful than others. For example, providing only human/chimp alignments will not lead to informative results. In fact, it is possible for vacuous or nearly-vacuous alignments (representing a reference sequence, but no appropriate informant species) to get a reasonably high score based solely on sequence composition of the reference sequence. (This arises from the generative nature of PhyloCSF’s underlying phylogenetic models.) Typically, with informative alignments, the substitution evidence is far stronger than the compositional biases; but in a vacuous alignment there is nothing else to go on. Therefore, it is generally advisable to provide PhyloCSF only with well-populated alignments that are likely to yield informative results about the patterns of codon substitution therein.
Regrettably, the algorithms for training the PhyloCSF parameters on different phylogenies are not yet available. We are still working on engineering a version suitable for public consumption. In the meantime, PhyloCSF does contain an experimental, simplified mode called the Omega Test for which it is much easier to provide parameters.
Suppose we have the alignment of a complete open reading frame, beginning at the start codon and ending immediately before the stop codon. This is the type of input one would typically provide to PAML. The above tal-AA.fa is such an example, and it’s included in the the distribution. Assuming we start in the main directory of the distribution, here’s how we’d run PhyloCSF on it:
$ ./PhyloCSF 12flies PhyloCSF_Examples/tal-AA.fa
PhyloCSF_Examples/tal-AA.fa decibans 297.6235
$ PhyloCSF thinks for a few seconds and reports that its model of protein-coding sequence evolution is strongly preferred over its non-coding model — by exactly 297.6235 decibans.
The first command line parameter 12flies tells PhyloCSF where to find the parameters it needs for the phylogeny of interest, which are stored in the PhyloCSF_Parameters subdirectory of the distribution. The name is actually a prefix to several other files found in that directory (12flies.nh, 12flies_coding.ECM and 12flies_noncoding.ECM). If you were analyzing mammals instead of flies, you would say 29mammals instead of 12flies.
Here’s a six-way alignment of the fifth coding exon of ALDH2, a highly conserved mammalian gene.
PhyloCSF_Examples/ALDH2.exon5.fa
>Human GTATTATGCCGGCTGGGCTGATAAGTACCACGGGAAAACCATCCCCATTGACGGAGACTTCTTCAGCTACACACGCCATGAACCTGTGGGGGTGTGCGGGCAGATCATTCCG >Mouse CTATTACGCTGGCTGGGCTGACAAGTACCATGGGAAAACCATTCCCATCGACGGCGACTTCTTCAGCTATACCCGCCATGAGCCTGTGGGCGTGTGTGGACAGATCATTCCG >Rat CTATTATGCTGGCTGGGCTGACAAGTACCACGGGAAAACCATTCCCATCGATGGCGACTTCTTCAGCTACACCCGCCACGAGCCTGTGGGCGTGTGTGGACAGATCATTCCG >Cow TTACTATGCCGGCTGGGCTGACAAATACCATGGGAAAACCATTCCCATTGACGGGGACTACTTCAGCTACACCCGCCATGAACCTGTGGGAGTGTGTGGGCAGATCATCCCA >Horse TTATTATGCTGGCTGGGCCGATAAGTACCATGGGAAAACCATTCCCATCGATGGGGACTTCTTCAGCTACACCCGCCATGAACCTGTGGGGGTGTGTGGGCAGATCATTCCG >Dog TTACTATGCTGGCTGGGCTGATAAGTACCATGGGAAAACCATTCCTATCGATGGGGACTTTTTCAGTTATACCCGCCATGAACCTGTCGGGGTGTGCGGGCAGATCATTCCA
If we try to run this like the last example:
$ ./PhyloCSF 29mammals PhyloCSF_Examples/ALDH2.exon5.fa
PhyloCSF_Examples/ALDH2.exon5.fa decibans -178.9246
$It gets a terrible score…what happened?? This exon has phase=1, i.e. it contains an extra nucleotide preceding the first complete codon. PhyloCSF doesn’t know that, though, so it evaluated the whole exon out-of-frame. We could trim this extra position off the alignment, but more conveniently, we can ask PhyloCSF to try all three reading frames and report the best:
$ ./PhyloCSF 29mammals PhyloCSF_Examples/ALDH2.exon5.fa --frames=3
PhyloCSF_Examples/ALDH2.exon5.fa decibans 218.2674 1 111
$There are two extra columns to the output, indicating the relative positions within the sequence of the region PhyloCSF is reporting. These positions are zero-based, inclusive. In this case, PhyloCSF has figured out that there’s an extra nucleotide at the beginning (position zero), so it’s reporting the score of the region from the second nucleotide (position one) through the end of the alignment. In this mode, it will always go to the end of the alignment; this will change later.
Now, suppose we didn’t even know the strand of transcription for this exon, in addition to not knowing the reading frame. We can ask for a six-frame search:
$ ./PhyloCSF 29mammals PhyloCSF_Examples/ALDH2.exon5.fa --frames=6
PhyloCSF_Examples/ALDH2.exon5.fa decibans 218.2674 1 111 +
$PhyloCSF tries the alignment (as given) in three frames, then reverse-complements it and tries three frames again, and finally reports the best of all six. In this case the ‘+’ indicates the best score was found in the alignment as given, not its reverse complement; it would be ‘-’ otherwise. The relative positions are always counting from the 5’-most position on the reported strand, so if the reported strand was ‘-’ then they would be counting from the end of the given alignment.
Search for the best ORF within a transcript alignment (boundaries, strand and/or reading frame unknown)
Now suppose we have an alignment of a (possibly incomplete) transcript model including one or more exons, and it may contain lengthy UTRs, or it may even be an entirely non-coding RNA. We can ask PhyloCSF to enumerate all possible ORFs above a given length, and report the best-scoring one.
For this example, we downloaded the complete mRNA of mouse Aldh2 (NM_009656.3) and its human (NM_000690.2) and dog (XM_848535.1) orthologs. Then we aligned them using EBI’s web server for MUSCLE. The results are in PhyloCSF_Examples/Aldh2.mRNA.fa (not reproduced here due to length). Note that we are using the mouse mRNA as the reference sequence, demonstrating the program’s agnosticism to the species order.
First, let’s see what happens if we run this more or less as in the preceding section, trying the complete alignment in three frames:
$ ./PhyloCSF 29mammals PhyloCSF_Examples/Aldh2.mRNA.fa --frames=3 --removeRefGaps --aa
PhyloCSF_Examples/Aldh2.mRNA.fa decibans 1910.4972 1 2206 FSSPPYLHRCEP*APATLLGAHT...
$We added two options to run it like this, --removeRefGaps to automatically remove alignment columns that are gapped in the reference (mouse) sequence, and --aa, which outputs the amino acid translation of the reported region (in the reference sequence). The translation has been truncated here as it is quite lengthy.
Note that although the alignment gets a high score overall, it contains some stop codons (the asterisks in the translation). This is because the mRNA includes UTRs. It doesn’t make a great deal of sense to include them in the analysis; if the UTRs were very lengthy, they could wash out the ORF and even lead to a negative overall score.
We can ask PhyloCSF to examine all start-to-stop ORFs within the transcript, and report the best:
$ ./PhyloCSF 29mammals PhyloCSF_Examples/Aldh2.mRNA.fa --orf=ATGStop --frames=3 --removeRefGaps --aa
PhyloCSF_Examples/Aldh2.mRNA.fa decibans 2013.9264 343 1899 MLRAALTTVRRGPRLSRLLSAAA...
$Note that the score improved by excluding the UTRs, there are no stop codons in the protein translation, and in fact PhyloCSF found the Aldh2 protein sequence exactly.
Furthermore, if we didn’t even known the strand of transcription for the alignment, we could use --frames=6.
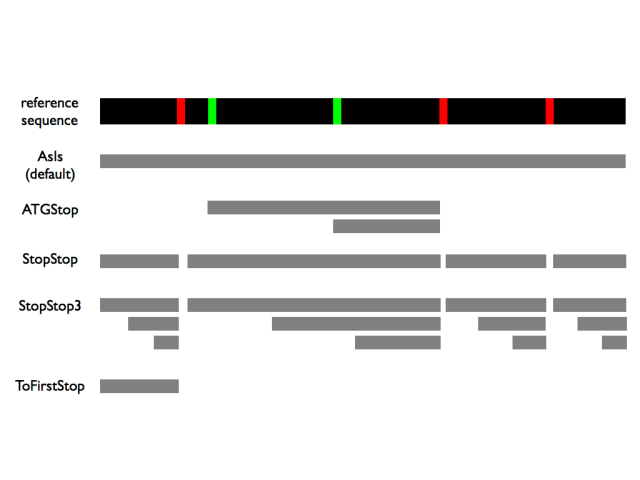
There are several possible settings for the --orf option, which enumerate possible ORFs in slightly different ways. The following schematic illustrates these settings, where the red bars indicate stop codons and the green bars indicate start codons in the reference sequence:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
This assumes that all the illustrated regions are above a minimum length, which can be specified with the --minCodons option. Also, it shows the ORFs only for one reading frame; it’s usually desirable to use an ORF search in three or six frames, in which case PhyloCSF will apply the requested strategy to each of the reading frames.
$ ./PhyloCSF --help usage: PhyloCSF.Linux.x86_64 parameter_set [file1 file2 ...] input will be read from stdin if no filenames are given.options:-h, --help show this help message and exit --strategy=mle|fixed|omega evaluation strategy (default mle)input interpretation:--files input list(s) of alignment filenames instead of individual alignment(s) --removeRefGaps automatically remove any alignment columns that are gapped in the reference sequence (nucleotide columns are removed individually; be careful about reading frame). By default, it is an error for the reference sequence to contain gaps --species=Species1,Species2,... hint that only this subset of species will be used in any of the alignments; this does not change the calculation mathematically, but can speed it upsearching multiple reading frames and ORFs:-f1|3|6, --frames=1|3|6 how many reading frames to search (default 1) --orf=AsIs|ATGStop|StopStop|StopStop3|ToFirstStop|FromLastStop|ToOrFromStop search for ORFs (default AsIs) --minCodons=INT minimum ORF length for searching over ORFs (default 25 codons) --allScores report scores of all regions evaluated, not just the maxoutput control:--bls include alignment branch length score (BLS) for the reported region in output --ancComp include ancestral sequence composition score in output --dna include DNA sequence in output, the part of the reference (first) sequence whose score is reported --aa include amino acid translation in output --debug print extra information about parameters and errors
PhyloCSF outputs a score, positive if the alignment is likely to represent a conserved coding region, and negative otherwise. This score has a precise theoretical interpretation: it quantifies how much more probable the alignment is under PhyloCSF’s model of protein-coding sequence evolution than under the non-coding model. As indicated in the output, this likelihood ratio is expressed in units of decibans. A score of 10 decibans means the coding model is 10:1 more likely than the non-coding model; 20 decibans, 100:1; 30 decibans, 1000:1; and so on. A score of -20 means the non-coding model is 100:1 more likely, and a score of 0 indicates the two models are equally likely.
This theoretical interpretation is nice to have, at least compared to a purely ad hoc score. However, if the likelihood ratio is, say, 1000:1 (30 decibans), we shouldn’t necessarily take those exact odds with our own money at stake, for the underlying models of sequence evolution make many simplifying assumptions that are not fully satisfied in reality. There are also more general caveats, such as:
- An alignment with a good score could represent an ancestral coding gene that recently “died”, and is now a pseudogene in the species of interest.
- An alignment with a poor score could be protein-coding in the species of interest, but not conserved in the others.
- The input alignment may not represent an appropriate orthologous region from some or all of the species. For example, a duplicated pseudogene could be aligned to its parent gene, or more generally, a complex orthology/paralogy situation may not be correctly resolved.
Therefore: please do not claim that anything “is” or “is not” protein-coding based solely on PhyloCSF. PhyloCSF provides one line of evidence either for or against the hypothesis that the input alignment represents a coding region, by examining evolutionary signatures. If you wish to make strong statements about whether you have found coding or non-coding regions, you should complement PhyloCSF with additional lines of evidence, such as homology searches to known protein sequence databases (e.g. BLASTX on nr or HMMER on Pfam).
It can also be helpful to compare the scores of novel regions with positive and negative control sets, e.g. cross-species alignments of orthologous mRNAs of known genes (positive) and long non-coding RNAs or even random non-coding regions (negative), generated using the same alignment pipeline.
Lastly, it is always, always, always a good idea to manually inspect at least some of the alignments for which PhyloCSF reports an interesting score, to ensure you are finding the types of examples you want.
The PhyloCSF score is a log-likelihood ratio of a given alignment under coding and non-coding evolutionary models. However, those models provide only an approximation of the likelihoods of actual alignments. Notably, the models assume statistical independence of codon sites, whereas in practice sites are not independent.
To address this, we defined PhyloCSF-Ψ, a region-length dependent score based on the empirical distributions of PhyloCSF scores in coding and non-coding regions of various lengths. PhyloCSF-Ψ estimates the relative likelihood that a raw PhyloCSF score would result from an actual coding or non-coding region of a particular length (Section 4.2 of Lin et al. 2011). This allows several things:
- Estimating how much more likely it is that a coding region of a particular length would get a particular score than a non-coding region. For example, a PhyloCSF-Ψ score of 20 decibans means that a coding region of that length is
$10^{20/10} = 100$ times more likely to get that PhyloCSF score than a non-coding region of that length.
- Setting a score threshold that is meaningful for regions of different lengths.
- Estimating a p-value. The formula for PhyloCSF-Ψ includes an approximation to the likelihood that a non-coding region of a particular length would get a specified PhyloCSF score. Integrating over all scores above some score provides a p-value for that score.
Computing PhyloCSF-Ψ requires six precomputed coefficients that depend on the particular multi-species whole-genome alignment being used. The “Psi” directory in the PhyloCSF installation contains files with these six coefficients for many common alignments and PhyloCSF parameters.
Note that the PhyloCSF-Ψ score will only be meaningful if the PhyloCSF score that is plugged into the PhyloCSF-Ψ computation was computed using a local alignment extracted from the same multispecies whole-genome alignment and using the same PhyloCSF parameters and strategy as were used to calculate the PhyloCSF-Ψ coefficients. For example, if using the PhyloCSF-Ψ coefficients for the 58 placental mammals alignment with mle strategy, the local alignment used as input to PhyloCSF must be extracted from the 100 vertebrates multispecies alignment; sequences from exactly those species not included among the 58 placental mammals most be removed, not more or fewer species; columns containing the final stop codon or gaps in the reference species should be removed; and the score must be computed using the (default) mle strategy with the 58mammals PhyloCSF parameters.
Warning: The PhyloCSF-Ψ score is not accurate in the case that the local alignment has very few substitutions, either because the phylogenetic branch length of the species present in the local alignment is very low (e.g., only a few closely-related species) or because there is very high nucleotide-level conservation, for example because of overlap with some other functional element. The reason is that although the branch length scaling used by PhyloCSF’s mle strategy protects against bias due to low branch length as regards whether the PhyloCSF score is positive or negative, low branch length (or low number of substitutions) tends to compress the score towards 0. The comparison to the precomputed score distributions of coding and non-coding regions will thus not be meaningful.
There are a few ways to use PhyloCSF to process a batch of alignments.
- Run it individually on each file, as above. This is easy but there is some inefficiency as the software must read the parameters for, and instantiate, large phylogenetic models each time you start it. If you do not specify a filename on the command line, PhyloCSF will expect an alignment piped through standard input.
- Provide several alignment filenames on the command line. PhyloCSF will proceed through each in turn, and the output is a reasonable tab-delimited format that includes the filename processed on each line.
- If you use the
--filesoption, then each filename supplied on the command line is assumed to refer to a file which itself contains a list of individual alignment files to process, one filename per line. - If you use the
--filesoption but do not supply any filenames on the command line, PhyloCSF will expect a list of alignment filenames piped through standard input, again one filename per line. In this mode PhyloCSF can be used on-line by a driver script: it reads an alignment filename from standard in, inputs and processes the alignment, reports the results (on standard output), and then input the next filename.
PhyloCSF will stop everything if it encounters a basic problem with the way an alignment is formatted (file not found, unknown species, invalid characters, etc.), as this suggests you need to make adjustments to your pipeline. For other errors, such as numerical convergence problems or no ORFs found in ORF search mode, PhyloCSF will report the problem but still continue on to the next alignment.
If you have many alignments to process, the best approach for multi-core parallelism is to run separate PhyloCSF processes on shards of the alignment set, by combining the batch techniques above with GNU Parallel or similar.
If you want the answer for one alignment ASAP, PhyloCSF can parallelize the search over different reading frames and ORFs by forking child processes to analyze each candidate region. As a corollary, this is only useful in conjunction with --frames and/or --orf on the command line. To enable this parallel mode:
opam install forkwork- Recompile PhyloCSF with
make FORKWORK=1 - Run PhyloCSF with
-p <# cores>on the command line.
The test suite can also be parallelized with make FORKWORK=1 test.